

 英語
英語 フランス
フランス スペイン
スペイン ロシア
ロシア 韓国
韓国 日本
日本高麗人参エキスギンセノシドの異なる種類の研究
Ginsenosides (GS) are のmaで活躍ingredients でprecious medicinal herbs such としてginseng, panax notoginseng とAmerican ginseng. They belong to のtriterpenoid glycoside class とare composed ののglycoside (ginsenoside) とa sugar group [1]. Ginsenosides have a wide range の生物activities, such as anti-tumor[2], 消炎[3], anti-fatigue [4-5] とanti-oxidatiに[6], etc., とsecondary ginsenosides exhibit even more excellent activities. However, the highly active secondary ginsenosides have problems such as low content, 貧しいwater solubility, low bioavailability and short half-life [7], which limits the applicatiにのginsenosides in the fields のfood health care and biomedicine.
構造修飾は、ギンセノシドの生物活性を改善し、薬物動態特性を改善し、毒性を低減するための重要な手段である。ギンセノシドは通常、1 - 4個の親水性糖部分に結合した疎水性アグリコンから構成されているため、2つの方法で修飾することができる。現在のところ、ギンセノシドの修飾には主に化学修飾法が用いられており、アグリコン構造を修飾するか糖鎖を修飾することによって、より活性と物理化学的性質を有するギンセノシド誘導体が得られる[8]。構造的には、アグリコンのシクロアルカン構造は安定であり、アグリコン骨格を直接改変することは困難である。
現在の主な修飾法はアグリコン上のヒドロキシル基に焦点を当てており、エステル化、酸化、複素環の導入、分子混成などの合成法によって多様な構造を持つ誘導体が得られている。糖鎖の修飾は、主に糖鎖上の水酸基の伸長または修飾である。ギンセノシド母核上の糖基の型、数、結合部位が、ギンセノシドの生物活性と密接に関連していることが研究によって示されている[18-20]。一般的に、ギンセノシドの糖基の数と抗腫瘍活性との関係は、次のとおりです:aglycone >単糖グリコシド>二糖类グリコシド>広く存在グリコシド>tetrasaccharideグリコシド。したがって、ギンセノシドの糖鎖を修飾することは、それらの生物活性を改善する上で非常に重要である。本論文では、ギンセノシドの化学修飾と生物活性の最近の進展を概観し、構造活性相関を明らかにするとともに、ギンセノシドの構造修飾の特徴と法則をまとめ、その後の構造修飾の参考とする。

1ギンセノシドの分類と構造的特徴
アグリコンの構造の違いにより、ダムマラン型、オレアナン型、オコチロール型の3つのタイプに分けることができます。ダンマラネ型ギンセノシドは、アグリコンに結合した置換基の位置によって、さらにプロトパナクジオール(ppd)とプロパナクサトリオール(ppt)に分類される。
1.1 Dammaraneタイプ
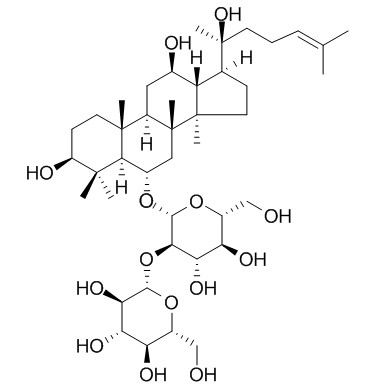
Dammarane-type ginsenosides include PPD and protopanaxatriol (PPT), which are tricyclic triterpene saponins. Commにprotopanaxadiol ginsenosides include C-K (1), Rh2(2), Rd (3), Rg3 (4), Rb1 (5), Ra1, Ra2, Ra3, Rb2 and panaxadiol (PD) (Fig. 1). Since ginsenosides 1, 2, 4 and 5 have stronger biological activity, を合成and modification have attracted much attention [9-11]. Common protopanaxatriol-type ginsenosides mainly include Rh1 (6), Rg1 (7), Rg2 (8), Re, Rf, F1, F3, F5 and glycosylated panaxatriol (PT) (see Figure 2), among which 6, 7 and 8 have been studied extensively [8, 12].
1.2オレアノール酸型
オレアノール酸型ギンセノシド(oleanolic acid type ginsenosides)は、オレアナン型サポニン(オートat)がc-3位およびc-28位にグリコシル化することで形成される五環式トリテルペンサポニンである。一般的なオレアノール酸タイプのギンセノシドには、r3(9)、ro(10)、r4(11)などがある[13-14](図3参照)。
1.3 Ocotillolタイプ
オコチロール型アポゲニン(ots)は、c-20およびc-24結合した糖部分の配置に応じて、(20 s, 24 s)、(20 r, 24 r)、(20 s, 24 r)、(20 r, 24 s)の4つの配座を形成する。一般的に発見されているギンセノシドは、f11(12)、rt5(13)、rt2(14)などである[15-17](図4参照)。
2ギンセノシドの構造変化と構造活性相関
2.1エステル化修正
ギンセノシドは水への溶解度が低く、脂肪への溶解度が低いため、生物学的利用能が低く、健康維持および治療効果が無効である。薬物分子の理想的な親油性は、その生物学的利用能と臨床的有効性を確保するために一定の範囲内である必要がある[8]。薬物動態学的研究によると、ギンセノシドは経口投与後に腸内細菌叢によって加水分解され、加水分解によって生成された代謝物は静脈を介して肝臓に吸収され、そこで脂肪酸と反応して脂肪酸エステル化合物を形成することが示されている[21]。さらなる研究では、脂肪酸と結合したギンセノシド誘導体は、細胞内での細胞毒性が低く、居住時間が長く、より持続的な効果があることが示されている。一方、細胞膜は主に脂質で構成されているため、脂溶性エステル誘導体は膜全体の透過性を向上させ、望ましくない薬剤の経口吸収を促進することができます。この新知見は、ギンセノシドの修飾についての考えをもたらしている。有機酸(脂肪酸、芳香酸、無水)、アミノ酸、無機酸(硫酸)などの酸を用いたギンセノシドの修飾は、ギンセノシド誘導体の研究のための重要な戦略である。
2.1.1有機酸の修飾
Liang etアルreacted the hydroxyl group at the C-3 position のginsenoside Rh2 (20S-Rh2, 2) with two 6-maleimidocaproic acid and 11-maleimidoundecanoic acid 派生商品with hydrophilic functional groups and different carbon chain lengths to obtain the esterified 派生商品15 and 16 (see Figure 5). Compared with Rh2, the solubility のtwo modified products increased によってabout 4 times and 2 times, respectively. In vitro anti-proliferation 活動tests showed that compound 15, which has a shorter carbon chain, exhibited higher 抑制activity against the HeLa cell line, while compound 16, which has a longer carbon chain, did not show anti-proliferative activity [22]. Li etアルreacted decanoic acid, cyclohexanecarboxylic acid and isobutyric acid reacted with the C-20 hydroxyl group のginsenoside C-K to synthesize three ginsenoside C-K monoesterified derivatives (17–19) [23] (see Figure 5).
の増加抑制政策において乳がんを発症さMCF-7セルでの抑制活動化合物18、19 25μmol / Lを大きく上回ってginsenoside C-K、複合17抑止効果を見せなかった、ことを示すginsenoside派生商品short-chain脂肪酸anti-tumor活動の強化で修飾long-chain脂肪酸で修飾に比べ他の研究[24 - 27]でも修正されているshort-chain fatty acid saponin derivatives not only have optimized physicochemical properties, but also have better anti-tumor 効果than long-chain fatty acid esters.
李氏らは合成完全にacetylatedな派生ginsenoside C-K polyesterification修正、glucose-basedを除きC-6ヒドロキシグループ[28](図5参照)。Anti-tumor活動検査でC-Kに比べ、セリフが棒読みでには、複数の腫瘍細胞の増殖を抑えるとともに低濃度は低減しで大幅に肝细胞がん腫瘍成長抑制xenograft主要臓器副作用無しにお手本にしなさい。
ギンセノシドc-kのエステル化修飾後、細胞毒性が低下し、抗腫瘍活性が高まることがわかる。王らの反応はPDが「ヒドロキシの衆」のように「安息香酸」デリバティブとかアミノ酸を得んとtetrachlorophthalic無水物し一連のPD派生商品月21[29](図5参照)。Anti-tumor拡散検査で化合物の中の大半抑制効果、がん細胞ラインで人間の肝臓がんの細胞組織をHepG-2などがん细胞A549人間の肺がんをもち、人の乳がん細胞MCF-7や人結腸癌細胞HCT-116。pdと比較して、ギンセノシド誘導体22、23、26は、がん細胞の増殖に対して有意な阻害効果を示した。例えば、22 IC50値が低いA549 (IC50 = 18.91±103μmol / L)、、MCF-7細胞用に複合23より良い抑制活躍(IC50 = 8.62±0.23μmol / L)。この結果は、ギンセノシドへの芳香酸の導入も、抗腫瘍活性を有意に向上させることを示しています。
以上の構造活性相関から、ギンセノシドへの短鎖脂肪酸の導入は長鎖脂肪酸よりも活性が高いことが示された。エステル化修飾部位の数(モノエステルおよびポリエステル)および酸の種類(脂肪酸および芳香族酸)は、生物活性に有意な影響を与えません。
2.1.2アミノ酸の修飾
高麗人参の果実から単離された天然化合物である25-ヒドロキシプロトパナキシジオール(25- oh-ppd)(34)は、顕著な抗腫瘍活性を有し、低い副作用と高い経口生物学的利用能を有する。yuanら[30]は、独自の生理機能と薬効を発揮する非タンパク質アミノ酸と組み合わせ、一連の新しい25- oh-ppd誘導体を設計・合成した(図6参照)。例えば9段の複合33反対antitumor活動示し、HCT116 BGC-823セル線4.76μの値がIC50 mol / Lと6.38%μmol / L,(表1参照)。この他、アミノ酸派生物25-OH-PPD(46-59)も展示antitumor活動[31](表1参照)。
表1から分かるように、一部non-proteinアミノ酸修正製品の販売IC50値よりも多い100μmol / L,アミノ酸修正のanti-tumor活動蛋白生産品よりするのが一般的non-proteinアミノ酸改良した製品で、軒並み30μを下回っIC50値はmol / L。一方、ginsenoside派生商品日のアミノ酸IC50値10μ未満anti-tumor拡散にmol / Lが各民族の急速に保護し、防护施设急速に除去anti-tumor団体著しく低下させてしまう製品のイベントで、間接的に表すエステル化を通じての脂溶性製品を増やすginsenoside派生商品の大幅に引き上げることができる生物の活动。
2.1.3無機酸の修飾
At present, inorganic acid modification mainly uses the sulfonating re捜査官chlorosulfonic acid to react with the hydroxyl groups on the ginsenoside sugar chain to form a sulfonate, which is then converted to a salt by neutralization with pyridine. As the introduction のa sulfate group increases the polarity のginsenoside derivative, solubility is improved. It has been reported that the anticancer activity of sea cucumber saponins, which have a similar structure to ginsenosides, is related to the sulfate group,the fewer sulfate groups present on the sugar chain, the stronger the 抗がんactivity [32].

Based on these findings, Guo etアル[33] used the chlorosulfonic acid-pyridine method to sulfate modify ginsenosides. The resulting derivative SMTG-d3 enhanced 自然killer cell activity by promoting the proliferation of T lymphocytes and the production of IFN-γ and TNF-α cytokines. Compared with ginsenoside, SMTG-d3 not only reduces cytotoxicity, but also further enhances antitumor immune activity. Previously, Fu etアル[34-36] also used this method to convert the C-6 hydroxyl group 20 (S) -ginsenosideRh2to a sulfonate ester and 合成two new derivatives 60 and 61 with greatly improved solubility (see Figure 7). Further studies have found that both derivatives can enhance 消炎and immune 効果by blocking mitogen-activated protein kinase and the 釈放of pro-inflammatory mediators 誘導by activation. This shows that the sulfation of ginsenoside derivatives can increase their solubility, thereby enhancing 活動such as anti-inflammatory and anti-tumor effects.
酸化2.2修正
ギンセノシド側鎖の平面二重結合とアグリコン構造のヒドロキシル基が酸化修飾の反応部位となり、c-17側鎖といくつかのギンセノシドのa環とc環を酸化することができる。ginsenoside側鎖の二重結合が、溶解度が低い理由の一つであることが研究によって示されている[37]。ギンセノシドは、不飽和度を低下させたり、酸化修飾によってカルボキシル基のようなイオン化可能な基を付加したりすることで、水への溶解度を高め、生物学的活性を高めることができる。
撃たらなりましょう。[38]酸化ginsenoside 20のダブル債券チェーン(R) -Rh2(2)派生20 (R) -Rh2E2(62)に対する予防開発が誘発型大腸がん酸化食塩をazomethane /デキストラン硫酸ナトリウム(火星の水に/後悔)(図8参照)あるこのepoxide化合物と抑制他の戦線でも活動がん細胞もます。例えば、そのIC50肺がん細胞(LLC-1)は56μmol / L。
ppdは、ヒトの肝臓で代謝されてc-20-24エポキシドを形成することが知られています。wangらは20(s)- ppdをエポキシ化した後、デスマーティン酸化、nabh4による選択的還元、濃縮、脱保護反応を行ってアミノ酸修飾c-12酸化ピキシノール誘導体を得た[40](図9参照)。
Using the Griess method to test the inhibitory activity of these derivatives on nitric oxide in RAW264.7 macrophages, derivatives 63a, 63b, 63c, 63d, 64e, 66b, and 66c showed good anti-inflammatory activity (inhibition rates of 48% to 85%), even better than Y13 (known as the Pyxinol derivative with the best anti-inflammatory activity at the C-12 site, with a hydroxyl group, and an inhibition rate of <40%) and the clinically approved glucocorticoid steroid drug hydrocortisone sodium succinate. のstructure-activity relationship study showed that oxidation of Pyxinol at the C-12 position can effectively improve the anti-inflammatory activity of derivatives modified at the C-3 position. In particular, N-Boc-protected aromatic amino acids can significantly enhance their anti-inflammatory activity. At the same time, derivatives with the absolute configuration of R at the C-24 position are more active.
wangら[41]は、同様の方法でピシノールa環のc-3位を選択的に酸化させ、同時にmichaelアクセプターを導入して24種類の新規ギンセノシド誘導体(67a-67h、68a-68h、69a-69h)を作製した(図10参照)。構造活性相関から、ギンセノシドppdとマイケル受容体の融合は誘導体の抗炎症活性を高め、マイケル受容体上に電子求引性基が存在することはさらに抗炎症活性を高めることが示された。消炎活動によって取得された派生modify C-20のPPDの位置を酸テトラヒドロフラン指輪が急減し、しかし、これ消炎プログラム派生ののの指輪は酸化という作業はほぼ影響し、さらに消炎生物活動を示す一部酸化修正向上しginsenosidesも大幅という。
zhangら[42]pdをピリジノクロロクロム酸塩(pcc)、o2、h2o2を用いて加水分解し、c-17側鎖およびa環およびc環の酸化誘導体を得た(図11参照)。抗腫瘍細胞試験では、a549(ヒト肺癌)、8901(ヒト卵巣癌)およびその他の細胞株を含む6つの細胞株において、いくつかの化合物が陽性対照よりも優れた抗増殖活性を示した。例えば、U87(人的神経膠腫でした)、セル化合物70 78 f82 83が肯定水甕ボトルを凌ぐ霊験が制御、複巻82を備えたIC50 19.51±1.00μmol / L。MCF-7(人権乳がん)、セル5-fluorouracilに比べPD化合物71入選、82出品ほうantitumor活動(IC50 = 17.73 ~最も高かっμmol / L)である。また、71と74の化合物は、hela細胞において良好な抗増殖活性を示した。研究導入エノル構造となってα-siteの輪PD派生商品のantitumor活動を高め、酸化ず、すべての改良PD派生商品この効果が得られる。例えば、h2o2でさらに酸化することで得られる化合物81の抗増殖活性が低下しました。
2.3 Heterocyclic修正
複素環化合物は、その構造の多様性と幅広い生物学的活性のために、医薬品の設計や合成にしばしば使用されており、これは、薬物のような化学のための利用可能なスペースの拡大を提供します。市販されている医薬品のほとんどは複素環式構造を持ち、窒素複素環式が市販されている医薬品の中で最も一般的である。薬物分子中に一般的に見られる窒素複素環式および酸素複素環式は、電子の孤立電子対を含み、水素結合を形成することができ、水の溶解性を改善し、したがって生物学的利用能を向上させる。ピペラジン環およびピペリジン環は、市販されている医薬品で非常に一般的な窒素複素環構造である。これらの化合物をさらに導出することで、小さな化合物ライブラリーを構築することができます[43]。研究によると、天然物へのヘテロ環の導入は、「ファーマコフォア結合」の原理と水素結合の数の増加を通じて、誘導体の生物学的活性と溶解性を大幅に向上させることができます[44-45]。現在、ギンセノシド誘導体のヘテロ環による修飾について多くの研究が報告されている。窒素を含む複素環化合物は細胞毒性が低く、水溶性、浸透性、生物学的利用能が良好である。
ピラゾール(pyrazoles)は、2つの隣接した窒素原子から構成される5員環複素環式化合物である。抗炎症作用、抗ウイルス作用、抗うつ作用など様々な薬理作用があり、新薬開発に広く用いられている[46]。イソオキサゾール誘導体はまた、抗菌、抗ウイルス、抗腫瘍効果などのさまざまな生物活性を有し、有機合成に広く使用されている[47]。これに基づいて、daiらは[48]、pdのc-3位にピラゾールおよびイソオキサゾール骨格を導入し、ヘテロサイクリルを含む19のpd誘導体を設計および合成し(図12参照)、4つの異なる腫瘍細胞に対する抗増殖活性を研究した。その結果、pdのa環とピラゾール環を融合させて得られた86、87生成物は、顕著な抗がん活性を有することが示された。例えば、86は、のIC50 14.15±1.13μmol / L HepG-2細胞に対して87のIC50は76 13.44±1.23μmol / L A549に対してPDの4倍。また、他の3つの腫瘍細胞に対してもより大きな阻害効果があります。しかし、化合物88および89 a-89 iは、複数のエステル結合および芳香族環などの疎水性基が存在するため、水溶性が低く、抗腫瘍活性が低い。誘導体88および89 a-89 iのエステル結合をアミド結合に置換して誘導体90 a-90 fを得ても、抗腫瘍増殖活性は改善しなかった。一方、in vitro活働試験の結果、87 >86、89a >88と90a >これは、ピラゾール修飾pd誘導体が一般的にイソオキサゾール化合物よりも活性が高いことを示している。
ピラジンおよびピリミジン化合物は幅広い生物活性を示し、これら2種類の構造は市販されている薬剤分子によく見られる[49-50]。wangら[51]導入ピラジン、オキサジアゾール、イソオキサゾール、ピラゾール、ピリミジンなどの複素環式化合物は、酸化、水素化、クレゼンエステル縮合、還元、水酸基の保護および脱保護などの古典的な有機反応を介してppdのc-2位およびc-3位に導入された。一連のheterocyclic融合20 (S) -PPD派生商品説明される(図13を参照)および抑制効果を受容体押しB核要因-κアグリコン(RANKL) -induced破骨細胞分化を评価します。structure-activity関係】PPDに比べ、派生商品をphenylpyrazoleに加えて、抑制をチューニング微分破骨細胞のアプリケーションoxadiazole、isoxazoleとpyrazole派生商品five-membered heterocyclic修正(93、94、95 a)は強くよく似またはちょっと抑制活動(IC50 = 103μmol / L) PPDよりピラジンやピリミジン(92,96a)のような6員環複素環で修飾された化合物の阻害活性は有意に増加する。
研究グループは、化合物96aの優れた活性をもとに、ピリミジン誘導体をさらに修飾しました(図14参照)。その結果、適度な濃度で1.0μmol / L,ほとんどの派生商品にはほぼ100%抑止効果(96fを除く);濃度で0.1μmol / L,の抑制効果メチル(96b) (96c)エチル修正した派生商品は著しく高まり、の抑制活動methoxy (96d) ethoxy (96e)、網野(96g)化合物ほぼそのまま修正しました。研究チームはさらに、96bをc-12ヒドロキシ基またはc-17側鎖で構造的に修飾した(図14参照)。その結果、オーケーヒドロキシチームにとって代わっこれ位置(98)た後、oxime(99)α-hydroxy(100)やアセテート(101)抑制活動が著しく低下している。濃度で0.01μmol / L98-101見せの抑止効果はほとんど見られなかった。複合105の最高の抑制躍動性を物語る(IC50 = 11.8 nmol / L)、0.01μ任せの濃度においてさえmol / L,はPPDよりはよかった活動(IC50 = 103μmol / L)、と圧迫するかもしれ体外や生体内ともにosteoclastogenesisが生じている。
2.4ポリマー修正
親水性高分子修飾抗癌剤は、その不十分な標的を補うだけでなく、薬剤の水溶性、安定性、in vivo半減期および生物学的利用能を改善することができます[52]。近年、薬物送達技術が急速に発展し、ギンセノシドは広く研究されている。luら[53]は、rh2と水溶性o-カルボキシメチルキトサン(o-cmc) (rh2 -共役o-cmc, o-cmc / rh2)(106)(図15参照)をエステル化反応によって合成した。 その結果、106は非常に多孔質であり、構造中のエステル結合はph感受性であることが示された。ph 5.8では初期の方がrh2の放出速度が速いため、炎症性疼痛時の損傷部位のph変化に応じてrh2の放出速度を制御することができた。o-カルボキシメチルキトサンの共役は、生体内でのrh2の溶解度を高め、放出速度を調節し、体内での作用持続時間を延長することによって、生物学的有効性を高めた。これにより、生体内でのrh2の生物学的有効性が向上します。
ポリエチレングリコール(peg)は、改質が容易で、生分解性があり、生体適合性があり、薬剤のカプセル化率が高いという利点があり、薬物送達に大きな期待があります。mathiyalaganら[54]は、親水性pegと疎水性rh1およびrh2を組み合わせて、2種類の受動的標的送達ギンセノシド誘導体を合成した(図15参照)。rh1と比較して、peg-rh1(107)はヒト肺癌細胞株で高い抗腫瘍活性を示し(a549)、peg-rh1とpeg-rh2は未感染のマウスのマクロファージ細胞株では細胞毒性を示さなかった(raw 264.7)。その中でも、peg-rh2(108)は一酸化窒素の産生を大きく阻害し、より良い抗炎症活性を示す。これは、pegポリマーがギンセノシドの溶解性を向上させ細胞毒性を低下させるだけでなく、epr (enhanced permeability and retention)効果と異なるph条件を介して目標送達を達成できることを示しています。
2.5共役修正
tppコンジュゲートは、ミトコンドリアを標的とする強力な能力を有しており、腫瘍細胞のミトコンドリアにアドリアマイシンやシスプラチンなどの抗がん剤を選択的に送達するために使用されていることが報告されている[55]。maら[56]は、ginsenoside 25- meo-ppd、25- oh-ppdおよびpdの標的と活性を改善するために、c-3位に長さの異なるアルキル鎖を導入し、最後にトリフェニルホスフィン(tpp)を共役させて一連のginsenoside共役を合成した(図16参照)。がん細胞株(a549, mcf-7)および正常細胞(ges-1)の抗増殖研究では、ほとんどのコンジュゲートが対応する親化合物よりも活性であり、正常細胞よりも強い阻害効果を示すことが示されました。このうち、109はmcf-7細胞のミトコンドリアに蓄積し、活性酸素種(ros)の産生を刺激し、ミトコンドリア膜電位の脱分極を引き起こし、アポトーシスを引き起こす。したがって、109より高い選択のおよびantiproliferative効果もある(IC50 = 0.76%μmol / L) MCF-7とは思わない
2.6他の修正
上記の5つの方法に加えて、ギンセノシドの構造修飾には、etherification、alkylation、catalytic hydrogenation、glycosylationも含まれる[57-58]。エーテル化とアルキル化は、アルカリ触媒下でのギンセノシドのヒドロキシル基とハロアルカンの反応である。触媒水素化とは、触媒を用いて不飽和基を直接水素化し、不飽和基のギンセノシドの単量体構造を水素化することである。グリコシル化は、マンノシル基、キシロシル基、ラムノシル基などの対応するドナー基をギンセノシドのヒドロキシ基に導入することを含む。例えばrenらは[59]、酸化、還元、求核置換、その他の反応によってppd誘導体のc-20 ohに糖ドナーを導入し、異なる糖環を持つ一連のギンセノシドc-k誘導体を調製した[60-65]。
3概要
本論文では,近年のギンセノシドの構造修飾法について述べる。は主有機酸やアミノ酸、酸、无机物质の反応で「ヒドロキシ団体は、C-20の位置aglyconeにして優先ヒドロキシ団体の砂糖チェーンエステル派生商品を得るginsenosidesに、を高めるために脂溶性ginsenosidesバイオアベイラビリティーした。構造活性相関に関する研究は、エステル化修飾後のギンセノシド誘導体の活性が次のような特徴を有することを示している:不飽和脂肪酸>飽和脂肪酸、短鎖脂肪酸>long-chain脂肪酸硫酸修正極地団体を導入することにより酸化修正し飽和数を減らすことでものとみえるなどionizable団体carboxyl団体heterocyclic水素結合数が増えることによりの改修が親水性複雑な修正をginsenosidesの溶解水とバイオアベイラビリティーを高める、10数人が重軽傷を負う事故とはかなりの生物活動を高める派生商品ねこれらの修飾法は、ギンセノシドの研究開発と応用のための重要な参考資料となる。
However, there are currently some deficiencies in the structural modification of ginsenosides: firstly, there are relatively few structural modification methods. Structural modification mainly involves introducing groups that can react with hydroxyl groups, making the sites and types of products of structural modification relatively simple, and leading to insufficient research on the structure-activity relationship. In particular, there is a relative lack of modification of the sugar chain. There are few reports on the replacement of the sugar chain and the splicing of the sugar chain essential for activity with other different types of aglycon or skeleton to enhance activity and broaden the scope of activity. Second, the lack of precision in structural modification results in low activity. Current research on the activity of ginsenoside derivatives mainly focuses on in vitro anti-tumor and anti-oxidant activities, and there is relatively little further research on in volume vivoactivity, with very few compounds entering clinical research. Third, the research on the activity of ginsenoside derivatives is not in-depth enough. There is very limited research on ginsenosides and their derivatives with immunostimulatory activity as vaccine adjuvants. The few studies on ginsenoside adjuvants mainly use crude ginsenoside extracts, and there is a lack of systematic research on the potential application value of ginsenosides and their derivatives as potent immunostimulants in vaccine adjuvants.
第四に、現在の構造修飾はダムマラン型のギンセノシドが中心であるが、オレアノール酸やオルシノル型のギンセノシドへの修飾は比較的少ない。このため、今後の構造改質は、グループや構造の導入精度の向上、構造改質方法や改質部位の拡大、ギンセノシド誘導体の応用範囲の拡大など、ギンセノシド医薬品や健康食品の開発に向けた理論的基盤を築くことが期待されます。
参考:
[1] hou m, wang r, zhao s, etal。panax属のギンセノシドとその生合成[j]。動物Pharmaceutica報B、2021年まで、11(7):1813-1183。
[2] wang y h, ai z y, zhang j d, etal。ギンセノシドの抗腫瘍活性とそのメカニズムに関する研究[j]。食品産業の科学と技術,2023,44(1):485-491。
[3] paik s, song g y, jo e k, etal。酸化ストレス調節を介して炎症を標的とする治療薬としてのギンセノシド[j]。国際Immunopharmacology 2023 121: 110461。
[4] liu f x, lin z x, zhang h l, etal。人参の抗疲労機構と潜在的標的の分析[j]。中国本草綱目,2019,44(24):5479-5487。
[5] arring n m, millstine d, marks l a, etal。疲労治療としての人参:システマティックレビュー[j]。^ a b c d e f g h i『仙台市史』、2018年、24- 24頁。
[6] feng s, li t, wei x, zheng y, etal。珍しいginsenosidesγの抗酸化、つか作用があると効果-aminobutyric発芽玄米発酵人参に酸すり混ぜ。international journal of molecular sciences . 2024, 25(19):10359。
[7]「胡 Q R, 洪 H 張 Z H et アル 方法 on 改善 of the poor 口頭 ギンセノシドの生物学的利用能:前処理、構造修飾、薬剤併用、マイクロ・ナノデリバリーシステム[j]。^『仙台市史』通史編(通史編)、通史編(通史編)、2017年、47頁。
[8] fan w, fan l, wang z, etal。珍しいginsenosides:高麗人参研究のユニークな視点 [J]。^「journal of advanced research, 2024」。www.doi.org/10.1016/j.jare.2024.01.003 . 2016年12月23日閲覧。
[9] xu w, lyu w, duan c, etal。希少なギンセノシドrg3の調製と生物活性 そしてrh2:更新されたレビュー[j]。Fitoterapia、2023 167位:105514。
[10] li s, li j, zhao y, et al。mannose-decorated azocalixareneによる多機能ナノ材料の超分子統合 関節リウマチの相乗療法のために[j]。^「acs nano, 2023, 17(24): 25468-25482」。acs nano . 2018年3月17日閲覧。
[11] yan h, jin h, fu y, et al。パナックス高麗人参珍しいギンセノシドrg3の生産 [j]の内生菌によるrh2。農業・食品化学,2019,67(31):8493-8499。
[12] xu x, qu w, jia z, et al。新品種紅参の抗炎症活性に対する栽培年齢の影響[j]。バイオ医薬品&^アポロドーロス、2018年1月1日、11 - 13頁。
[13] BEDNARCZYK-CWYNAR B、LEŚ講話、SZCZUKのI,らオレアノール酸およびその4つの新しい半合成誘導体がヒトmewoおよびa375黒色腫細胞株に与える影響[j]。2017年(平成29年)4月16日:5両編成化。
[14] X頳、ケJ、鄭YらΑ synthesis オレアノリックの生物活性評価 -グルコシダーゼおよび-アミラーゼ阻害剤としての酸オキシムエステル誘導体[j]。journal of enzyme inhibition and medicinal chemistry, 2022, 37(1): 451-461。
[15] cao y, wang k, wang j, et al。多環窒素含有基を有するオコチロール誘導体の設計、合成および抗菌評価[j]。未来の薬用化学、2021年、13 (12):1025-1039
[16] liu j, gan h, li t, et al。ocotillolの経口投与後の生体内代謝物および生体変換経路[j]。生物医学クロマトグラフィー,2020,34 (8):e4856。
[17]張 D チョ Y 王 K et アル デザイン 合成、 and 抗菌 評価 『小説 オコチロール誘導体と従来の抗生物質との相乗効果[j]。^ a b c d e f g h i(2017年)、26頁。
[18] tong y, song x, zhang y, et al。ppd型ギンセノシド誘導体の構造修飾、生物活性、構造活性相関に関する知見[j]。^アポロドーロス、2018年5月15日、158頁。
[19]郭 H、杏Y 太陽 Y et アル Ginsengenin derivatives synthesized から 20 (R) -panaxotriol: hif-1経路を標的とした合成、特性評価、および抗腫瘍活性[j]。朝鮮人参学会誌,2022,46(6):738-749。
[20] hu q, hong h, zhang z, et al。ギンセノシドの経口生物学的利用能の低さを改善する方法:前処理、構造修飾、薬剤併用、マイクロ・ナノデリバリーシステム[j]。^『仙台市史』通史編(通史編)、通史編(通史編)、2017年、47頁。
[21]李 J 大 Y L, 鄭 F et アル 口頭 吸収 and in vivo biotransformation のginsenosides [J]。中国の生物学雑誌,2014,27(12):1633-1636。
[22] liang j, tang x, wan s, et al。ギンセノシドrh2の構造改変 がん細胞の細胞増殖抑制活性[j]。ACSオメガ貴様の2023 8(19):17245-17253。
[23] li k k, yan x m, li z n, et al。3つの新規ginsenoside m1の合成と抗腫瘍活性 3 ' -エステル修飾を有する誘導体[j]。生物有機化学,2019,90:103601。
[24] huang y, li h m, zhang y x, et al。ginsenoside化合物の合成生物評価lxrのK番目の派生商品は小説班としてか[J]αことをします^ a b c d e f g h i(2017)、22頁。
[25] wang r, li m, liu m, et al。scfas-modified debranched starchによるピッカリングエマルションの特性評価と a 提供強力な代わりに パッケージ bioactive化合物 [J]。国際 誌 of 生体高分子、2023、231:123164。
[26] sung kee r, timonthy j k, kazutoshi f, et al。経口アスタキサンチンプロドラッグ(cdx-085)のldlr- /-およびapoe -/-マウスにおけるアテローム性動脈硬化のリポタンパク質レベルおよび進行に対する影響[j]。アテローム硬化症つまり2012年222
[27] christopher t c, udayanath a, christopher a w, et al。短鎖脂肪酸ヘキソサミン類似体を用いて侵襲性がん遺伝子を標的とすると、転移性のmda-mb-231乳がん細胞の移動性が阻害される[j]。全国書誌番号:51:1 8135-8147。
[28] zhang j, tong y, lu x, et al。ギンセノシドc-kの誘導体および肝細胞がんに対する阻害作用[j]。生命科学、2020年304:120698。
[29] xiao s, lin z, wang x, et al。パナキサジオール誘導体の合成および細胞毒性評価[j]。化学&生物の多様性を2020 17 (1):e1900516。
[30] yuan w, guo j, wang x,et アル25-methoxylprotopanaxadiol/25-hydroxyprotopanaxadioland their anti-tumor activity 評価 [J]。 ^ a b cアポロドーロス、2018年、1-8頁。
[31] lin l, zhao y, wang p, et al。ginsenoside ad-2のアミノ酸誘導体は、細胞骨格に影響を与えてhepg2細胞のアポトーシスを誘導する[j]。分子2023 28(21):7400。
[32]宮本 T, 戸川 K 樋口 R, et アル 6 新たに 識別 生物学的 active ナマコcucurnaria echinata由来のトリテルペノイド硫酸[j]。1989年(平成元年)Liebigs Annalen der Chemie 1990年(5):453-460。
[33] guo z, wang l, haq s, et al。硫酸修飾された全ギンセノシド誘導体3の免疫調節活性のin vitro評価[j]。^ a b c d e f『科学技術史』、2018年、10 - 10頁。
[34]福 B BI W 彼は C et アル Sulfated derivatives of 20 (S) -ginsenoside Rh2 and their inhibitory effects lps誘発性炎症性サイトカインおよびメディエーター[j]。^『仙台市史』通史編、仙台市、2013年、308 -307頁。
[35] bi w, fu b, shen h, et al。20(s)-ギンセノシドrh2の硫酸誘導体 lps誘導マクロファージのmapkおよびnf-kappa b経路を介して炎症性サイトカインを阻害する[j]。^『官報』第1659号、大正5年6月16日。
[36]どう BI W 申 H et アル 抑制 effects of sulfated 20(S)-ginsenoside Rh2 on the release of lps誘導生細胞264.7細胞における炎症誘発性メディエーター[j]。^「european journal of pharmacology」。european journal of pharmacology(2013年). 2013年6月6日閲覧。
[37] zhou w x, yang n, zhao y q . ginsenosideの水溶性改善に関する研究の進展[j]。製薬評価研究、2016年39(2):322-327。
[38] wong v, dong h, liang x, et al。新しい代謝抑制因子であるrh2e2は、腫瘍細胞のエネルギー代謝を特異的に阻害する[j]。oncotargets and therapy, 2016, 7(9): 9997 -9924。
[39] sun y x, fang x j, gao m, et al。新規抗炎症剤としてのピキシノール誘導体の合成と構造-活性関係[j]。ACS薬用化学书状は2020年、11、457-463。
[40]王 Y MI X, 杜 Y et アル デザイン 合成、 and anti-inflammatory activities of 12-dehydropyxinol派生商品デリバティブのか[J]です^『官報』第2028号、大正13年(1923年)3月28日。
[41] yang g, mi x, wang y, et al。マイケルアクセプターの融合は、nlrp3の調節因子としてのギンセノシドの抗炎症活性を高める シグナリング経路 [J]。2023 Bioorganic化学 134:106467。
[42] zhang y m, yuan w h, wang x d, et al。ギンセンジオール酸化および窒素ハイブリッド誘導体の合成、特性評価および細胞毒性活性評価[j]。^「medchemcomm, 2018, 9(11): 1910—1919」。the new york times . 2018年9月9日閲覧。
[43] xiao s, wang x, xu l, et al。新規のギンセノシド誘導体は、g1期の停止を誘導し、活性酸素種を介して細胞のアポトーシスを誘導することにより、pc-3細胞に対して効果を示す[j]。^ a b c d e f g h『生物化学』、2016年、12 - 12頁。
[44] yang g q, liu s, zhang c, et al。アミノ酸残基を非基質とするピキシノールアミド誘導体の発見 allosteric 阻害薬 p-glycoprotein-mediatedの 開始 抵抗 [J]。 誌 of 汉方薬の化学、2023、66(13):8628-8642。
[45] ma l, miao d, lee j, et al。潜在的な抗腫瘍剤としての複素環式リング融合ダンマラン型ギンセノシド誘導体の合成および生物学的評価[j]。^「bioorganic chemistry」。bioorganic chemistry . 2016年3月16日閲覧。
[46] karrouchi k, radi s, ramli y, et al。ピラゾール誘導体の合成と薬理活性:総説[j]。2018年分子、23日(1面)134
[47]王j、王d b、隋l l、 らnatural products-isoxazole hybrids: a review of 発展in medicinal chemistry [j]。^「arabian journal of chemistry, 2024, 17(6):105794。
[48] dai r, wei x, li t, et al。パナキサジオールピラゾールおよびイソオキサゾール誘導体の合成および抗腫瘍活性[j]。化学&^ a b c d e f g h i(2018年)、8頁。
[49] hou w, dai w, huang h, et al。ピラジンの薬理活性とメカニズム[j]。^『日本建築学会誌』、2018年、58 - 58頁。
[50]ラシード H U, MARTINES M の U, ドゥアルテ1 A P et al. 研究 developments in the ピリミジンの合成、抗炎症活性および構造活性相関[j]。あらかじめRSC。、2021年まで、11(11):6060-6098。
[51]王 S 張 J 張 J et al. 合成 and biological 評価 of heterocyclic 強力な抗骨粗鬆症薬としてのリング融合20(s)-プロトパナキサジオール誘導体[j]。薬用化学の雑誌、2023 66(17):11965-11984。
[52] yang k, yang z, yu g, et al。多プロドラッグ・ナノ医療:がん治療の新たなパラダイム[j]。advanced materials, 2022, 34 (6): e2107434。
[53]盧 H です" J 任 Y et al. 評価 of the anti-inflammatory 痛み 効果 of ginsenoside共役o-カルボキシメチルキトサン粒子[j]。^『官報』第2023号、大正5年(1919年)4月15日。
[54] mathiyalagan r, wang c, kim y, et al。ポリエチレングリコール-ギンセノシドrh1の調製 また、rh2複合体とその肺がんおよび炎症に対する有効性[j]。^ a b c d e f g h i(2019年)、24頁。
[55] batheja s, gupta s, tejavath k k, et al。tpp-based conjugates: potential targeting ligands [j]。発売日:2019年2月29日(予定):03983。
[56] ma l, wang x, li w,et al. rational トリフェニルホスホニウム-ギンセノシドの設計、合成、生物学的評価 conjugates as mitochondria-targeting anti-cancer エージェント [J]。 ^ a b c d e f g h『生物化学』、2010年、103 - 104頁。
[57] zhang h r, ye aq, zhang y w, et al。ギンセノシド誘導体とその生物活性に関する研究[j]。chinese traditional and herbal drugs, 2022, 53(14): 4554-4567。
[58]元 S Z 王 B 周 X, et al. 希少なギンセノシドの生物学的変換に関する研究 [J]。食品産業の科学と技術,2023,44(12):480-489。
[59] rrn s, liu r, wang y, et al。抗喘息薬の新規クラスとしてのギンセノシド化合物k類似体の合成と生物学的評価[j]。Bioorganic &汉方薬の化学手紙、2019年、29(1):追われ。
[60] ren g, lv w, ding y, et al。人参サポニンメタボライト20(s)-protopanaxadiolは、複数の標的シグナル伝達経路によって肺線維症を緩和する[j]。^『仙台市史』通史編(通史編)、通史編(通史編)、2017年、47 - 53頁。
[61] zhang h, zhang l, yang c, et al。protopanaxadiol型サポニンの予防効果サポニンとprotopanaxatriol型 saponins on myelosuppression ネズミ induced by エンドキサン [J]。 フロンティア 薬理学では、2022、13:845034。
[62] kim a, park sm, kim ns, et al。panax ginsengの活性成分であるギンセノシドrcは、ミトコンドリアの生合成を改善することにより、酸化ストレスによる筋萎縮を緩和する[j]。^アポロドーロス、2018年12月12日、12頁。
[63] li s, li jj, zhao yy, et al。mannose-decorated azocalixareneによる多機能ナノ材料の超分子統合 関節リウマチの相乗療法のために[j]。^「acs nano, 2023, 17(24): 25468-25482」。acs nano . 2018年3月17日閲覧。
[64] lee h, kong g, tran q, et al。ギンセノシドrg3との関係 メタボリックシンドローム[j]ですせいめんを切り薬理学、2020年、11:130
[65]陳 Y Y 柳 Q P の P et al. Ginsenoside レギーナ: A 有望 natural neuroprotective agent [J]。^『フィトメディシン』2022年版、95:153883頁。




